 3D Periodic Editor 3D Molecular And Reaction Editor Expert Editor Microsoft Quantum Editor EMSL Aerosol Workshop Editor Manual 3D Periodic Editor 3D Molecular And Reaction Editor Expert Editor Microsoft Quantum Editor EMSL Aerosol Workshop Editor Manual
3D Periodic Editor 3D Molecular And Reaction Editor Expert Editor Microsoft Quantum Editor EMSL Aerosol Workshop Editor Manual 3D Periodic Editor 3D Molecular And Reaction Editor Expert Editor Microsoft Quantum Editor EMSL Aerosol Workshop Editor Manual Results from an EMSL Arrows Calculation
| EMSL Arrows is a revolutionary approach to materials and chemical simulations that uses NWChem and chemical computational databases to make materials and chemical modeling accessible via a broad spectrum of digital communications including posts to web APIs, social networks, and traditional email. |
Molecular modeling software has previously been extremely complex, making it prohibative to all but experts in the field, yet even experts can struggle to perform calculations. This service is designed to be used by experts and non-experts alike. Experts can carry out and keep track of large numbers of complex calculations with diverse levels of theories present in their workflows. Additionally, due to a streamlined and easy-to-use input, non-experts can carry out a wide variety of molecular modeling calculations previously not accessible to them.
The id(s) for emsiles = CC(Cl)(C)C theory{dft} xc{b3lyp} basis{6-311++G(2d,2p)} solvation_type{COSMO} ^{0} are: 80978
Use id=% instead of esmiles to print other entries.
mformula = C4Cl1H9
iupac = 2-chloro-2-methylpropane
PubChem = 10486
PubChem LCSS = 10486
cas = 507-20-0
synonyms = 2-Chloro-2-methylpropane; 507-20-0; tert-Butyl chloride; Propane, 2-chloro-2-methyl-; Trimethylchloromethane; tert-Butylchloride; Chlorotrimethylmethane; 2-Chloroisobutane; t-Butylchloride; 2-Methyl-2-chloropropane; 2-Methyl-2-propyl chloride; NSC 6527; EINECS 208-066-4; JN2YO95TZ0; BRN 1730872; CCRIS 9388; DTXSID2023937; AI3-30754; NSC-6527; BUTYL CHLORIDE, TERT-; DTXCID803937; TERT-BUTYL CHLORIDE [MI]; EC 208-066-4; 4-01-00-00288 (Beilstein Handbook Reference); tButyl chloride; 2Chloroisobutane; tertButylchloride; 2Chloro2methylpropane; 2Methyl2chloropropane; Butyl chloride, tert; 2Methyl2propyl chloride; Propane, 2chloro2methyl; Chloro2methyl propane, 2; PROPANE,2-CHLORO,2-METHYL TERT.BUTYL,CHLORIDE; 208-066-4; inchi=1/c4h9cl/c1-4(2)3-5/h4h,3h2,1-2h; inchi=1/c4h9cl/c1-4(2,3)5/h1-3h; T-BUTYL CHLORIDE; Tertiary-butyl chloride; 2-Chloro-2-methyl-propane; UNII-JN2YO95TZ0; 2-Chloro-2-methyl propane; tertbutyl chloride; t-BuCl; tert.butyl chloride; MFCD00000816; tert-C4H9Cl; tert.-butyl chloride; Propane, 2-chloro-2-methyl; 2-chloranyl-2-methyl-propane; CHEMBL346997; NSC6527; 2-Chloro-2-methylpropane, 99%; WLN: GX1&1&1; STR03102; Tox21_200192; AKOS000121885; NCGC00248555-01; NCGC00257746-01; CAS-507-20-0; DB-050414; tert-Butylchloride 2-Chloro-2-methylpropane; NS00006377; EN300-29211; A828266; Q209343; F0001-1327; PROPANE,2-CHLORO,2-METHYL TERT.BUTYL,CHLORIDE; Z281778956; 2-Chloro-2-methylpropane, puriss. p.a., >=99.0% (GC)
Search Links to Other Online Resources (may not be available):
- Google Structure Search
- EPA CompTox Database
- Chemical Entities of Biological Interest (ChEBI)
- NIH ChemIDplus - A TOXNET DATABASE
- The Human Metabolome Database (HMDB)
- OECD eChemPortal
- Google Scholar
+==================================================+
|| Molecular Calculation ||
+==================================================+
Id = 80978
NWOutput = Link to NWChem Output (download)
Datafiles:
homo-restricted.cube-100581-2025-4-26-10:38:46 (download)
lumo-restricted.cube-100581-2025-4-26-10:38:46 (download)
dft-b3lyp-189260.cosmo.xyz-100581-2025-4-26-10:38:46 (download)
mo_orbital_nwchemarrows-2025-5-16-0-38-189910.out-134891-2025-5-17-13:37:42 (download)
image_resset: api/image_reset/80978
Calculation performed by Eric Bylaska - aqe
Numbers of cpus used for calculation = 119
Calculation walltime = 228.500000 seconds (0 days 0 hours 3 minutes 48 seconds)
+----------------+
| Energetic Data |
+----------------+
Id = 80978
iupac = 2-chloro-2-methylpropane
mformula = C4Cl1H9
inchi = InChI=1S/C4H9Cl/c1-4(2,3)5/h1-3H3
inchikey = NBRKLOOSMBRFMH-UHFFFAOYSA-N
esmiles = CC(Cl)(C)C theory{dft} xc{b3lyp} basis{6-311++G(2d,2p)} solvation_type{COSMO} ^{0}
calculation_type = ovc
theory = dft
xc = b3lyp
basis = 6-311++G(2d,2p)
charge,mult = 0 1
energy = -618.147511 Hartrees
enthalpy correct.= 0.129637 Hartrees
entropy = 77.859 cal/mol-K
solvation energy = -1.762 kcal/mol solvation_type = COSMO
Sitkoff cavity dispersion = 2.148 kcal/mol
Honig cavity dispersion = 6.441 kcal/mol
ASA solvent accesible surface area = 257.655 Angstrom2
ASA solvent accesible volume = 236.599 Angstrom3
+-----------------+
| Structural Data |
+-----------------+
JSmol: an open-source HTML5 viewer for chemical structures in 3D
Number of Atoms = 14
Units are Angstrom for bonds and degrees for angles
Type I J K L M Value
----------- ----- ----- ----- ----- ----- ----------
1 Stretch C1 C2 1.52317
2 Stretch C1 H6 1.08853
3 Stretch C1 H7 1.09258
4 Stretch C1 H8 1.08853
5 Stretch C2 Cl3 1.86872
6 Stretch C2 C4 1.52317
7 Stretch C2 C5 1.52319
8 Stretch C4 H9 1.08853
9 Stretch C4 H10 1.08852
10 Stretch C4 H11 1.09259
11 Stretch C5 H12 1.08851
12 Stretch C5 H13 1.09257
13 Stretch C5 H14 1.08852
14 Bend C2 C1 H6 111.16513
15 Bend C2 C1 H7 109.25124
16 Bend C2 C1 H8 111.16298
17 Bend H6 C1 H7 108.32407
18 Bend H6 C1 H8 108.52877
19 Bend H7 C1 H8 108.32224
20 Bend C1 C2 Cl3 106.86542
21 Bend C1 C2 C4 111.94833
22 Bend C1 C2 C5 111.94253
23 Bend Cl3 C2 C4 106.86552
24 Bend Cl3 C2 C5 106.86817
25 Bend C4 C2 C5 111.94624
26 Bend C2 C4 H9 111.16344
27 Bend C2 C4 H10 111.16266
28 Bend C2 C4 H11 109.25630
29 Bend H9 C4 H10 108.52557
30 Bend H9 C4 H11 108.32096
31 Bend H10 C4 H11 108.32550
32 Bend C2 C5 H12 111.16655
33 Bend C2 C5 H13 109.24510
34 Bend C2 C5 H14 111.17141
35 Bend H12 C5 H13 108.32245
36 Bend H12 C5 H14 108.52735
37 Bend H13 C5 H14 108.32130
38 Dihedral C1 C2 C4 H9 -56.17632
39 Dihedral C1 C2 C4 H10 -177.18834
40 Dihedral C1 C2 C4 H11 63.31510
41 Dihedral C1 C2 C5 H12 56.16541
42 Dihedral C1 C2 C5 H13 -63.32259
43 Dihedral C1 C2 C5 H14 177.18782
44 Dihedral Cl3 C2 C1 H6 -60.50433
45 Dihedral Cl3 C2 C1 H7 -179.99739
46 Dihedral Cl3 C2 C1 H8 60.51314
47 Dihedral Cl3 C2 C4 H9 60.50472
48 Dihedral Cl3 C2 C4 H10 -60.50730
49 Dihedral Cl3 C2 C4 H11 179.99614
50 Dihedral Cl3 C2 C5 H12 -60.51408
51 Dihedral Cl3 C2 C5 H13 179.99792
52 Dihedral Cl3 C2 C5 H14 60.50834
53 Dihedral C4 C2 C1 H6 56.17677
54 Dihedral C4 C2 C1 H7 -63.31629
55 Dihedral C4 C2 C1 H8 177.19424
56 Dihedral C4 C2 C5 H12 -177.19571
57 Dihedral C4 C2 C5 H13 63.31629
58 Dihedral C4 C2 C5 H14 -56.17329
59 Dihedral C5 C2 C1 H6 -177.18548
60 Dihedral C5 C2 C1 H7 63.32146
61 Dihedral C5 C2 C1 H8 -56.16800
62 Dihedral C5 C2 C4 H9 177.18794
63 Dihedral C5 C2 C4 H10 56.17592
64 Dihedral C5 C2 C4 H11 -63.32064
+-----------------+
| Eigenvalue Data |
+-----------------+
Id = 80978
iupac = 2-chloro-2-methylpropane
mformula = C4Cl1H9
InChI = InChI=1S/C4H9Cl/c1-4(2,3)5/h1-3H3
smiles = CC(Cl)(C)C
esmiles = CC(Cl)(C)C theory{dft} xc{b3lyp} basis{6-311++G(2d,2p)} solvation_type{COSMO} ^{0}
theory = dft
xc = b3lyp
basis = 6-311++G(2d,2p)
charge = 0
mult = 1
solvation_type = COSMO
twirl webpage = TwirlMol Link
image webpage = GIF Image Link
Eigevalue Spectra
---- ---- 65.99 eV
---- ----
----------
---- ----
-- -- -- -
--- -- ---
- - - - --
-- -- -- -
----------
6 - - - -
-- -- -- -
6 - - - -
---- ----
--- -- ---
- - - - --
----------
---- ----
-- -- -- -
--- -- ---
-- -- -- -
6 - - - -
9 - - - -
6 - - - -
6 - - - -
---- ----
-- -- -- -
7 - - - -
14 - - - -
9 - - - -
- - - - -- LUMO= -0.29 eV
HOMO= -8.41 eV + + + + ++
+++ ++ +++
+++ ++ +++
++++++++++
++++ ++++
++++++++++
-23.77 eV ++++++++++
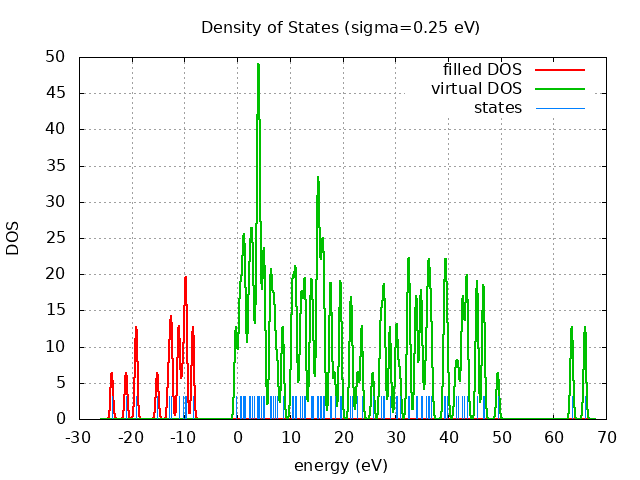
spin eig occ ---------------------------- restricted -23.77 2.00 restricted -21.09 2.00 restricted -19.17 2.00 restricted -19.17 2.00 restricted -15.18 2.00 restricted -12.94 2.00 restricted -12.48 2.00 restricted -12.48 2.00 restricted -11.08 2.00 restricted -11.08 2.00 restricted -10.32 2.00 restricted -9.79 2.00 restricted -9.79 2.00 restricted -9.73 2.00 restricted -8.41 2.00 restricted -8.41 2.00 restricted -0.29 0.00 restricted -0.19 0.00 restricted 0.47 0.00 restricted 0.47 0.00 restricted 0.75 0.00 restricted 1.08 0.00 restricted 1.08 0.00 restricted 1.32 0.00 restricted 1.48 0.00 restricted 1.48 0.00 restricted 2.17 0.00 restricted 2.19 0.00 restricted 2.52 0.00 restricted 2.52 0.00 restricted 2.87 0.00 restricted 2.90 0.00 restricted 2.90 0.00 restricted 3.28 0.00 restricted 3.78 0.00 restricted 3.78 0.00 restricted 4.00 0.00 restricted 4.01 0.00 restricted 4.04 0.00 restricted 4.05 0.00 restricted 4.21 0.00 restricted 4.21 0.00 restricted 4.22 0.00 restricted 4.51 0.00 restricted 4.86 0.00 restricted 5.12 0.00 restricted 5.14 0.00 restricted 5.14 0.00 restricted 6.33 0.00 restricted 6.35 0.00 restricted 6.36 0.00 restricted 6.88 0.00 restricted 6.89 0.00 restricted 7.21 0.00 restricted 7.66 0.00 restricted 8.66 0.00 restricted 8.66 0.00 restricted 10.45 0.00 restricted 10.45 0.00 restricted 10.63 0.00 restricted 11.07 0.00 restricted 11.10 0.00 restricted 11.10 0.00 restricted 11.96 0.00 restricted 12.25 0.00 restricted 12.25 0.00 restricted 12.72 0.00 restricted 12.87 0.00 restricted 12.87 0.00 restricted 13.96 0.00 restricted 13.96 0.00 restricted 14.25 0.00 restricted 14.41 0.00 restricted 15.24 0.00 restricted 15.24 0.00 restricted 15.38 0.00 restricted 15.38 0.00 restricted 15.42 0.00 restricted 15.69 0.00 restricted 16.00 0.00 restricted 16.01 0.00 restricted 16.34 0.00 restricted 16.40 0.00 restricted 16.40 0.00 restricted 17.67 0.00 restricted 17.67 0.00 restricted 17.76 0.00 restricted 18.50 0.00 restricted 19.60 0.00 restricted 19.60 0.00 restricted 19.63 0.00 restricted 21.51 0.00 restricted 21.52 0.00 restricted 21.78 0.00 restricted 22.85 0.00 restricted 23.63 0.00 restricted 23.63 0.00 restricted 25.73 0.00 restricted 27.25 0.00 restricted 27.25 0.00 restricted 27.70 0.00 restricted 27.91 0.00 restricted 27.91 0.00 restricted 28.98 0.00 restricted 28.98 0.00 restricted 30.25 0.00 restricted 30.27 0.00 restricted 30.87 0.00 restricted 32.40 0.00 restricted 32.40 0.00 restricted 32.66 0.00 restricted 32.66 0.00 restricted 33.88 0.00 restricted 33.88 0.00 restricted 34.13 0.00 restricted 34.84 0.00 restricted 34.84 0.00 restricted 35.05 0.00 restricted 35.85 0.00 restricted 36.25 0.00 restricted 36.35 0.00 restricted 36.35 0.00 restricted 36.80 0.00 restricted 36.80 0.00 restricted 39.29 0.00 restricted 39.45 0.00 restricted 39.45 0.00 restricted 39.83 0.00 restricted 39.83 0.00 restricted 41.48 0.00 restricted 41.96 0.00 restricted 42.74 0.00 restricted 42.74 0.00 restricted 43.02 0.00 restricted 43.56 0.00 restricted 43.56 0.00 restricted 43.58 0.00 restricted 45.52 0.00 restricted 45.52 0.00 restricted 45.57 0.00 restricted 46.65 0.00 restricted 46.79 0.00 restricted 46.79 0.00 restricted 49.44 0.00 restricted 63.50 0.00 restricted 63.50 0.00 restricted 65.99 0.00 restricted 65.99 0.00
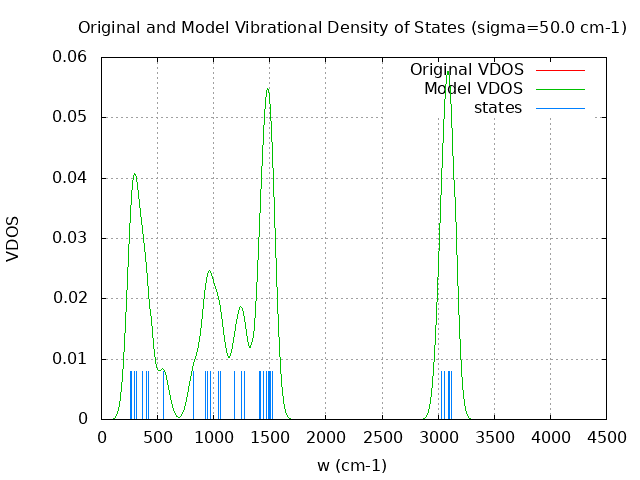
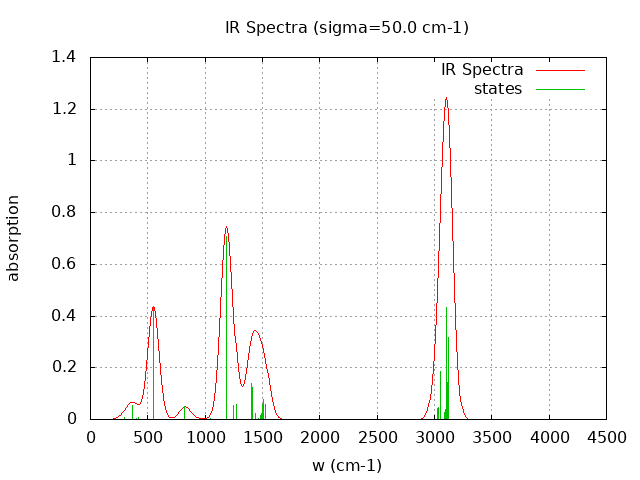
+----------------------------------------+ | Vibrational Density of States Analysis | +----------------------------------------+ Total number of frequencies = 42 Total number of negative frequencies = 0 Number of lowest frequencies = 8 (frequency threshold = 500 ) Exact dos norm = 36.000 Generating vibrational DOS Generating model vibrational DOS to have a proper norm 10.00 36.01 8.00 36.00 50.00 36.00 8.00 36.00 100.00 35.99 7.99 36.00 Generating IR Spectra Writing vibrational density of states (DOS) to vdos.dat Writing model vibrational density of states (DOS_FIXED) to vdos-model.dat Writing IR spectra to irdos.dat Temperature= 298.15 zero-point correction to energy = 76.666 kcal/mol ( 0.122175) vibrational contribution to enthalpy correction = 78.980 kcal/mol ( 0.125862) vibrational contribution to Entropy = 11.920 cal/mol-k hindered rotor enthalpy correction = 0.000 kcal/mol ( 0.000000) hindered rotor entropy correction = 0.000 cal/mol-k
DOS sigma = 10.000000
- vibrational DOS enthalpy correction = 0.125864 kcal/mol ( 78.981 kcal/mol)
- model vibrational DOS enthalpy correction = 0.125854 kcal/mol ( 78.975 kcal/mol)
- vibrational DOS Entropy = 0.000019 ( 11.930 cal/mol-k)
- model vibrational DOS Entropy = 0.000019 ( 11.920 cal/mol-k)
- original gas Energy = -618.147511 (-387893.417 kcal/mol)
- original gas Enthalpy = -618.017874 (-387812.068 kcal/mol, delta= 0.000)
- unajusted DOS gas Enthalpy = -618.017873 (-387812.067 kcal/mol, delta= 0.001)
- model DOS gas Enthalpy = -618.017882 (-387812.073 kcal/mol, delta= -0.005)
- original gas Entropy = 0.000124 ( 77.859 cal/mol-k,delta= 0.000)
- unajusted DOS gas Entropy = 0.000124 ( 77.869 cal/mol-k,delta= 0.010)
- model DOS gas Entropy = 0.000124 ( 77.859 cal/mol-k,delta= -0.000)
- original gas Free Energy = -618.054868 (-387835.282 kcal/mol, delta= 0.000)
- unadjusted DOS gas Free Energy = -618.054871 (-387835.284 kcal/mol, delta= -0.002)
- model DOS gas Free Energy = -618.054875 (-387835.287 kcal/mol, delta= -0.005)
- original sol Free Energy = -618.057675 (-387837.044 kcal/mol)
- unadjusted DOS sol Free Energy = -618.057678 (-387837.046 kcal/mol)
- model DOS sol Free Energy = -618.057683 (-387837.048 kcal/mol)
DOS sigma = 50.000000
- vibrational DOS enthalpy correction = 0.125900 kcal/mol ( 79.003 kcal/mol)
- model vibrational DOS enthalpy correction = 0.125900 kcal/mol ( 79.003 kcal/mol)
- vibrational DOS Entropy = 0.000019 ( 12.185 cal/mol-k)
- model vibrational DOS Entropy = 0.000019 ( 12.185 cal/mol-k)
- original gas Energy = -618.147511 (-387893.417 kcal/mol)
- original gas Enthalpy = -618.017874 (-387812.068 kcal/mol, delta= 0.000)
- unajusted DOS gas Enthalpy = -618.017837 (-387812.045 kcal/mol, delta= 0.024)
- model DOS gas Enthalpy = -618.017837 (-387812.045 kcal/mol, delta= 0.024)
- original gas Entropy = 0.000124 ( 77.859 cal/mol-k,delta= 0.000)
- unajusted DOS gas Entropy = 0.000124 ( 78.124 cal/mol-k,delta= 0.265)
- model DOS gas Entropy = 0.000124 ( 78.124 cal/mol-k,delta= 0.265)
- original gas Free Energy = -618.054868 (-387835.282 kcal/mol, delta= 0.000)
- unadjusted DOS gas Free Energy = -618.054956 (-387835.337 kcal/mol, delta= -0.056)
- model DOS gas Free Energy = -618.054956 (-387835.337 kcal/mol, delta= -0.056)
- original sol Free Energy = -618.057675 (-387837.044 kcal/mol)
- unadjusted DOS sol Free Energy = -618.057764 (-387837.099 kcal/mol)
- model DOS sol Free Energy = -618.057764 (-387837.099 kcal/mol)
DOS sigma = 100.000000
- vibrational DOS enthalpy correction = 0.126002 kcal/mol ( 79.067 kcal/mol)
- model vibrational DOS enthalpy correction = 0.126016 kcal/mol ( 79.076 kcal/mol)
- vibrational DOS Entropy = 0.000021 ( 13.120 cal/mol-k)
- model vibrational DOS Entropy = 0.000021 ( 13.138 cal/mol-k)
- original gas Energy = -618.147511 (-387893.417 kcal/mol)
- original gas Enthalpy = -618.017874 (-387812.068 kcal/mol, delta= 0.000)
- unajusted DOS gas Enthalpy = -618.017735 (-387811.980 kcal/mol, delta= 0.088)
- model DOS gas Enthalpy = -618.017720 (-387811.971 kcal/mol, delta= 0.097)
- original gas Entropy = 0.000124 ( 77.859 cal/mol-k,delta= 0.000)
- unajusted DOS gas Entropy = 0.000126 ( 79.059 cal/mol-k,delta= 1.200)
- model DOS gas Entropy = 0.000126 ( 79.077 cal/mol-k,delta= 1.218)
- original gas Free Energy = -618.054868 (-387835.282 kcal/mol, delta= 0.000)
- unadjusted DOS gas Free Energy = -618.055298 (-387835.552 kcal/mol, delta= -0.270)
- model DOS gas Free Energy = -618.055292 (-387835.548 kcal/mol, delta= -0.266)
- original sol Free Energy = -618.057675 (-387837.044 kcal/mol)
- unadjusted DOS sol Free Energy = -618.058105 (-387837.314 kcal/mol)
- model DOS sol Free Energy = -618.058100 (-387837.310 kcal/mol)
Normal Mode Frequency (cm-1) IR Intensity (arbitrary)
----------- ---------------- ------------------------
1 -0.000 0.374
2 -0.000 0.613
3 -0.000 0.190
4 -0.000 0.311
5 0.000 0.978
6 0.000 0.380
7 262.470 0.029
8 265.840 0.044
9 294.930 0.777
10 296.920 0.866
11 312.560 0.180
12 368.510 6.638
13 397.710 0.553
14 414.870 0.899
15 548.430 54.580
16 823.710 6.089
17 925.480 0.188
18 947.870 0.186
19 973.190 0.017
20 1046.560 0.244
21 1063.920 0.196
22 1184.560 88.832
23 1249.300 6.596
24 1273.180 7.353
25 1407.970 17.440
26 1412.400 15.655
27 1442.000 2.934
28 1469.310 0.504
29 1484.240 2.047
30 1490.910 3.016
31 1497.480 7.283
32 1508.890 9.530
33 1527.250 7.337
34 3029.070 5.169
35 3033.550 5.768
36 3053.130 23.369
37 3090.140 3.214
38 3097.050 4.948
39 3102.030 54.381
40 3117.500 18.132
41 3118.860 22.492
42 3122.490 39.666
No Hindered Rotor Data
+-------------------------------------+
| Reactions Contained in the Database |
+-------------------------------------+
Reactions Containing INCHIKEY = NBRKLOOSMBRFMH-UHFFFAOYSA-N
Reactionid Erxn(gas) Hrxn(gas) Grxn(gas) delta_Solv Grxn(aq) ReactionType Reaction
20962 4.698 3.461 4.204 1.993 6.197 AB + CD --> AD + BC "ClC(C)(C)C + O --> CC(C)(C)O + Cl"
20922 -1.255 -1.751 -1.690 1.436 -0.254 AB + CD --> AD + BC "CC(C)(C)I + Cl --> CC(C)(C)Cl + I"
20921 -1.255 -1.751 -1.690 1.436 -0.254 AB + CD --> AD + BC "CC(C)(C)I + Cl --> CC(C)(C)Cl + I"
20920 -1.255 -1.751 -1.690 1.436 -0.254 AB + CD --> AD + BC "CC(C)(C)I + Cl --> CC(C)(C)Cl + I"
20919 -1.255 -1.751 -1.690 1.436 -0.254 AB + CD --> AD + BC "CC(C)(C)I + Cl --> CC(C)(C)Cl + I"
20726 -0.748 -1.239 -1.178 1.436 0.258 AB + CD --> AD + BC "CC(C)(C)I + Cl --> CC(C)(C)Cl + I"
20725 -0.748 -1.239 -1.178 1.436 0.258 AB + CD --> AD + BC "CC(C)(C)I + Cl --> CC(C)(C)Cl + I"
20724 -0.748 -1.239 -1.178 1.436 0.258 AB + CD --> AD + BC "CC(C)(C)I + Cl --> CC(C)(C)Cl + I"
20723 -0.748 -1.239 -1.178 1.436 0.258 AB + CD --> AD + BC "CC(C)(C)I + Cl --> CC(C)(C)Cl + I"
20409 431.106 426.557 417.619 -152.976 264.643 AB --> A + B "ClC(C)(C)C --> C[C](C)C ^{-1} + [Cl] ^{1}"
20408 431.106 426.557 417.619 -152.976 264.643 AB --> A + B "ClC(C)(C)C --> C[C](C)C ^{-1} + [Cl] ^{1}"
20406 148.050 145.823 134.658 -141.119 -6.461 AB --> A + B "CC(C)(C)Cl --> C[C+](C)C + [Cl-]"
20405 148.050 145.823 134.658 -141.119 -6.461 AB --> A + B "CC(C)(C)Cl --> C[C+](C)C + [Cl-]"
20374 4.983 3.335 4.811 1.749 6.560 AB + CD --> AD + BC "ClC(C)(C)C + Ch3OH --> Cl + O(C(C)(C)C)C"
17085 2.803 2.243 2.303 0.000 2.303 AB + CD --> AD + BC "CC(C)(C)I theory{pspw4} + Cl theory{pspw4} --> CC(C)(C)Cl theory{pspw4} + I theory{pspw4}"
17084 2.803 2.243 2.303 0.000 2.303 AB + CD --> AD + BC "CC(C)(C)I theory{pspw4} + Cl theory{pspw4} --> CC(C)(C)Cl theory{pspw4} + I theory{pspw4}"
17083 2.803 2.243 2.303 0.000 2.303 AB + CD --> AD + BC "CC(C)(C)I theory{pspw4} + Cl theory{pspw4} --> CC(C)(C)Cl theory{pspw4} + I theory{pspw4}"
17082 2.803 2.243 2.303 0.000 2.303 AB + CD --> AD + BC "CC(C)(C)I theory{pspw4} + Cl theory{pspw4} --> CC(C)(C)Cl theory{pspw4} + I theory{pspw4}"
17070 -0.746 -1.230 -1.255 1.396 0.141 AB + CD --> AD + BC "CC(C)(C)I + Cl --> CC(C)(C)Cl + I"
17069 -0.746 -1.230 -1.255 1.396 0.141 AB + CD --> AD + BC "CC(C)(C)I + Cl --> CC(C)(C)Cl + I"
17068 -0.746 -1.230 -1.255 1.396 0.141 AB + CD --> AD + BC "CC(C)(C)I + Cl --> CC(C)(C)Cl + I"
17067 -0.746 -1.230 -1.255 1.396 0.141 AB + CD --> AD + BC "CC(C)(C)I + Cl --> CC(C)(C)Cl + I"
5997 431.104 426.548 417.695 -152.936 264.759 AB --> A + B "ClC(C)(C)C --> C[C](C)C ^{-1} + [Cl] ^{1}"
5996 431.104 426.548 417.695 -152.936 264.759 AB --> A + B "ClC(C)(C)C --> C[C](C)C ^{-1} + [Cl] ^{1}"
2792 148.048 145.814 134.735 -141.079 -6.344 AB --> A + B "CC(C)(C)Cl --> C[C+](C)C + [Cl-]"
2110 4.695 3.452 4.280 2.033 6.313 AB + CD --> AD + BC "ClC(C)(C)C + O --> CC(C)(C)O + Cl"
1782 4.981 3.326 4.887 1.789 6.676 AB + CD --> AD + BC "ClC(C)(C)C + Ch3OH --> Cl + O(C(C)(C)C)C"
1591 4.981 3.326 4.887 1.789 6.676 AB + CD --> AD + BC "ClC(C)(C)C + Ch3OH --> Cl + O(C(C)(C)C)C"
1278 148.048 145.814 134.735 -141.079 -6.344 AB --> A + B "CC(C)(C)Cl --> C[C+](C)C + [Cl-]"
96 4.695 3.451 4.279 2.033 6.312 AB + CD --> AD + BC "ClC(C)(C)C + O --> CC(C)(C)O + Cl"
58 4.981 3.320 4.859 1.869 6.728 AB + CD --> AD + BC "ClC(C)(C)C + Ch3OH --> HCl + O(C(C)(C)C)C"
All requests to Arrows were successful.
KEYWORDs -
reaction: :reaction
chinese_room: :chinese_room
molecule: :molecule
nmr: :nmr
predict: :predict
submitesmiles: :submitesmiles
nosubmitmissingesmiles
resubmitmissingesmiles
submitmachines: :submitmachines
useallentries
nomodelcorrect
eigenvalues: :eigenvalues
frequencies: :frequencies
nwoutput: :nwoutput
xyzfile: :xyzfile
alleigs: :alleigs
allfreqs: :allfreqs
reactionenumerate:
energytype:[erxn(gas) hrxn(gas) grxn(gas) delta_solvation grxn(aq)] :energytype
energytype:[kcal/mol kj/mol ev cm-1 ry hartree au] :energytype
tablereactions:
reaction: ... :reaction
reaction: ... :reaction
...
:tablereactions
tablemethods:
method: ... :method
method: ... :method
...
:tablemethods
:reactionenumerate
rotatebonds
xyzinput:
label: :label
xyzdata:
... xyz data ...
:xyzdata
:xyzinput
submitHf: :submitHf
nmrexp: :nmrexp
findreplace: old text | new text :findreplace
listnwjobs
fetchnwjob: :fetchnwjob
pushnwjob: :pushnwjob
printcsv: :printcsv
printeig: :printeig
printfreq: :printfreq
printxyz: :printxyz
printjobinfo: :printjobinfo
printnwout: :printnwout
badids: :badids
hup_string:
database:
table:
request_table:
listallesmiles
queuecheck
This software service and its documentation were developed at the Environmental Molecular Sciences Laboratory (EMSL) at Pacific Northwest National Laboratory, a multiprogram national laboratory, operated for the U.S. Department of Energy by Battelle under Contract Number DE-AC05-76RL01830. Support for this work was provided by the Department of Energy Office of Biological and Environmental Research, and Department of Defense environmental science and technology program (SERDP). THE SOFTWARE SERVICE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE SERVICE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE SERVICE.