 3D Periodic Editor 3D Molecular And Reaction Editor Expert Editor Microsoft Quantum Editor EMSL Aerosol Workshop Editor Manual 3D Periodic Editor 3D Molecular And Reaction Editor Expert Editor Microsoft Quantum Editor EMSL Aerosol Workshop Editor Manual
3D Periodic Editor 3D Molecular And Reaction Editor Expert Editor Microsoft Quantum Editor EMSL Aerosol Workshop Editor Manual 3D Periodic Editor 3D Molecular And Reaction Editor Expert Editor Microsoft Quantum Editor EMSL Aerosol Workshop Editor Manual Results from an EMSL Arrows Calculation
| EMSL Arrows is a revolutionary approach to materials and chemical simulations that uses NWChem and chemical computational databases to make materials and chemical modeling accessible via a broad spectrum of digital communications including posts to web APIs, social networks, and traditional email. |
Molecular modeling software has previously been extremely complex, making it prohibative to all but experts in the field, yet even experts can struggle to perform calculations. This service is designed to be used by experts and non-experts alike. Experts can carry out and keep track of large numbers of complex calculations with diverse levels of theories present in their workflows. Additionally, due to a streamlined and easy-to-use input, non-experts can carry out a wide variety of molecular modeling calculations previously not accessible to them.
The id(s) for emsiles = ClC/C=C/Cl theory{ccsd(t)} xc{unknown} basis{6-311++G(2d,2p)} solvation_type{COSMO} ^{0} theory_base{dft} xc_base{b3lyp} basis_base{6-311++G(2d,2p)} are: 44042
Use id=% instead of esmiles to print other entries.
mformula = C3Cl2H4
iupac = ClC/C=C/Cl
PubChem =
PubChem LCSS =
Search Links to Other Online Resources (may not be available):
- Google Structure Search
- EPA CompTox Database
- Chemical Entities of Biological Interest (ChEBI)
- NIH ChemIDplus - A TOXNET DATABASE
- The Human Metabolome Database (HMDB)
- OECD eChemPortal
- Google Scholar
+==================================================+
|| Molecular Calculation ||
+==================================================+
Id = 44042
NWOutput = Link to NWChem Output (download)
Datafiles:
image_resset: api/image_reset/44042
Calculation performed by Eric Bylaska - arrow14.emsl.pnl.gov
Numbers of cpus used for calculation = 32
Calculation walltime = 2533.500000 seconds (0 days 0 hours 42 minutes 13 seconds)
+----------------+
| Energetic Data |
+----------------+
Id = 44042
iupac = ClC/C=C/Cl
mformula = C3Cl2H4
inchi = InChI=1S/C3H4Cl2/c4-2-1-3-5/h1-2H,3H2/b2-1+
inchikey = UOORRWUZONOOLO-OWOJBTEDSA-N
esmiles = ClC/C=C/Cl theory{ccsd(t)} xc{unknown} basis{6-311++G(2d,2p)} solvation_type{COSMO} ^{0} theory_base{dft} xc_base{b3lyp} basis_base{6-311++G(2d,2p)}
calculation_type = ovc
theory = ccsd(t)
xc = unknown
basis = 6-311++G(2d,2p)
charge,mult = 0 1
energy = -1035.813683 Hartrees
enthalpy correct.= 0.069237 Hartrees
entropy = 78.674 cal/mol-K
solvation energy = -3.363 kcal/mol solvation_type = COSMO
Sitkoff cavity dispersion = 2.147 kcal/mol
Honig cavity dispersion = 6.437 kcal/mol
ASA solvent accesible surface area = 257.487 Angstrom2
ASA solvent accesible volume = 253.989 Angstrom3
+-----------------+
| Structural Data |
+-----------------+
JSmol: an open-source HTML5 viewer for chemical structures in 3D
Number of Atoms = 9
Units are Angstrom for bonds and degrees for angles
Type I J K L M Value
----------- ----- ----- ----- ----- ----- ----------
1 Stretch C1 Cl2 1.83583
2 Stretch C1 C3 1.48478
3 Stretch C1 H6 1.08586
4 Stretch C1 H7 1.08482
5 Stretch C3 C4 1.32474
6 Stretch C3 H8 1.08177
7 Stretch C4 Cl5 1.74406
8 Stretch C4 H9 1.07951
9 Bend Cl2 C1 C3 111.14542
10 Bend Cl2 C1 H6 104.70338
11 Bend Cl2 C1 H7 106.11715
12 Bend C3 C1 H6 112.10688
13 Bend C3 C1 H7 112.57541
14 Bend H6 C1 H7 109.74005
15 Bend C1 C3 C4 121.78012
16 Bend C1 C3 H8 117.65657
17 Bend C4 C3 H8 120.56307
18 Bend C3 C4 Cl5 123.30053
19 Bend C3 C4 H9 123.89373
20 Bend Cl5 C4 H9 112.80567
21 Dihedral C1 C3 C4 Cl5 -178.85857
22 Dihedral C1 C3 C4 H9 1.03383
23 Dihedral Cl2 C1 C3 C4 -115.18813
24 Dihedral Cl2 C1 C3 H8 64.98677
25 Dihedral C4 C3 C1 H6 128.01416
26 Dihedral C4 C3 C1 H7 3.70311
27 Dihedral Cl5 C4 C3 H8 0.96152
28 Dihedral H6 C1 C3 H8 -51.81094
29 Dihedral H7 C1 C3 H8 -176.12199
30 Dihedral H8 C3 C4 H9 -179.14608
+-----------------+
| Eigenvalue Data |
+-----------------+
Id = 44042
iupac = ClC/C=C/Cl
mformula = C3Cl2H4
InChI = InChI=1S/C3H4Cl2/c4-2-1-3-5/h1-2H,3H2/b2-1+
smiles = ClC/C=C/Cl
esmiles = ClC/C=C/Cl theory{ccsd(t)} xc{unknown} basis{6-311++G(2d,2p)} solvation_type{COSMO} ^{0} theory_base{dft} xc_base{b3lyp} basis_base{6-311++G(2d,2p)}
theory = ccsd(t)
xc = unknown
basis = 6-311++G(2d,2p)
charge = 0
mult = 1
solvation_type = COSMO
twirl webpage = TwirlMol Link
image webpage = GIF Image Link
Eigevalue Spectra
---------- 14.22 eV
---- ----
----------
----------
---- ----
---- ----
--- -- ---
- - - - --
- - - - --
- - - - --
---- ----
--- -- ---
---- ---- LUMO= 1.02 eV
HOMO= -10.25 eV ++++++++++
+++ ++ +++
++++++++++
++++ ++++
++++++++++
++++ ++++
++++++++++
++++++++++
++++++++++
++++++++++
-32.02 eV ++++++++++
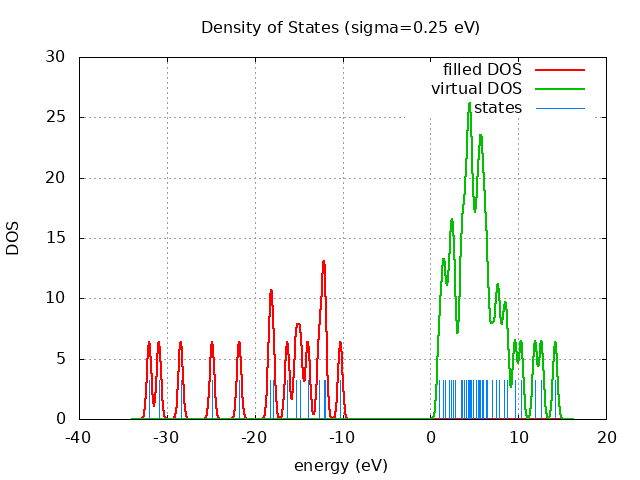
spin eig occ ---------------------------- restricted -32.02 2.00 restricted -30.91 2.00 restricted -28.43 2.00 restricted -24.86 2.00 restricted -21.77 2.00 restricted -18.27 2.00 restricted -17.97 2.00 restricted -16.30 2.00 restricted -15.29 2.00 restricted -14.80 2.00 restricted -13.96 2.00 restricted -12.65 2.00 restricted -12.15 2.00 restricted -12.05 2.00 restricted -10.25 2.00 restricted 1.02 0.00 restricted 1.43 0.00 restricted 1.63 0.00 restricted 2.07 0.00 restricted 2.37 0.00 restricted 2.53 0.00 restricted 2.80 0.00 restricted 3.44 0.00 restricted 3.60 0.00 restricted 3.89 0.00 restricted 4.06 0.00 restricted 4.28 0.00 restricted 4.45 0.00 restricted 4.50 0.00 restricted 4.66 0.00 restricted 4.87 0.00 restricted 5.17 0.00 restricted 5.37 0.00 restricted 5.59 0.00 restricted 5.70 0.00 restricted 5.92 0.00 restricted 5.99 0.00 restricted 6.33 0.00 restricted 6.47 0.00 restricted 7.02 0.00 restricted 7.52 0.00 restricted 7.81 0.00 restricted 8.35 0.00 restricted 8.73 0.00 restricted 9.62 0.00 restricted 10.33 0.00 restricted 11.91 0.00 restricted 12.64 0.00 restricted 14.22 0.00
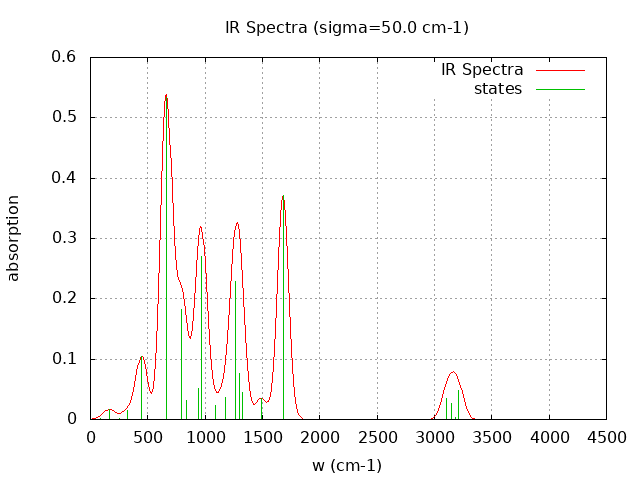
+----------------------------------------+ | Vibrational Density of States Analysis | +----------------------------------------+ Total number of frequencies = 27 Total number of negative frequencies = 0 Number of lowest frequencies = 5 (frequency threshold = 500 ) Exact dos norm = 21.000 Generating vibrational DOS Generating model vibrational DOS to have a proper norm 10.00 21.00 5.00 21.00 50.00 20.96 4.96 21.00 100.00 20.76 4.76 21.00 Generating IR Spectra Writing vibrational density of states (DOS) to vdos.dat Writing model vibrational density of states (DOS_FIXED) to vdos-model.dat Writing IR spectra to irdos.dat Temperature= 298.15 zero-point correction to energy = 39.225 kcal/mol ( 0.062508) vibrational contribution to enthalpy correction = 41.078 kcal/mol ( 0.065462) vibrational contribution to Entropy = 11.081 cal/mol-k hindered rotor enthalpy correction = 0.000 kcal/mol ( 0.000000) hindered rotor entropy correction = 0.000 cal/mol-k
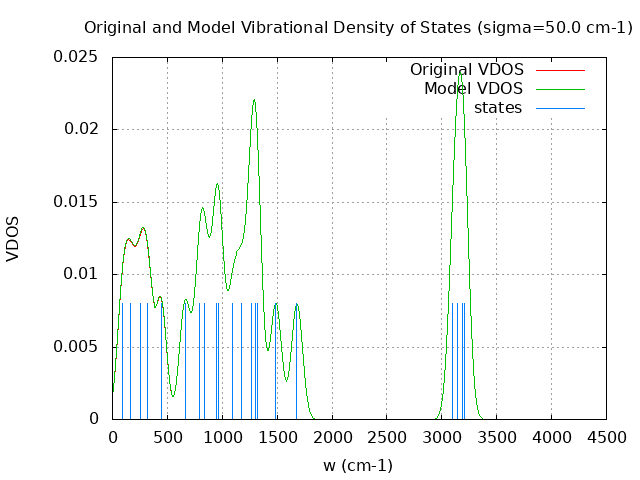
DOS sigma = 10.000000
- vibrational DOS enthalpy correction = 0.065463 kcal/mol ( 41.079 kcal/mol)
- model vibrational DOS enthalpy correction = 0.065465 kcal/mol ( 41.080 kcal/mol)
- vibrational DOS Entropy = 0.000018 ( 11.101 cal/mol-k)
- model vibrational DOS Entropy = 0.000018 ( 11.105 cal/mol-k)
- original gas Energy = -1035.813683 (-649982.894 kcal/mol)
- original gas Enthalpy = -1035.744446 (-649939.447 kcal/mol, delta= 0.000)
- unajusted DOS gas Enthalpy = -1035.744445 (-649939.447 kcal/mol, delta= 0.001)
- model DOS gas Enthalpy = -1035.744443 (-649939.445 kcal/mol, delta= 0.002)
- original gas Entropy = 0.000125 ( 78.674 cal/mol-k,delta= 0.000)
- unajusted DOS gas Entropy = 0.000125 ( 78.694 cal/mol-k,delta= 0.020)
- model DOS gas Entropy = 0.000125 ( 78.698 cal/mol-k,delta= 0.024)
- original gas Free Energy = -1035.781826 (-649962.904 kcal/mol, delta= 0.000)
- unadjusted DOS gas Free Energy = -1035.781835 (-649962.909 kcal/mol, delta= -0.005)
- model DOS gas Free Energy = -1035.781835 (-649962.909 kcal/mol, delta= -0.005)
- original sol Free Energy = -1035.787185 (-649966.266 kcal/mol)
- unadjusted DOS sol Free Energy = -1035.787194 (-649966.272 kcal/mol)
- model DOS sol Free Energy = -1035.787193 (-649966.272 kcal/mol)
DOS sigma = 50.000000
- vibrational DOS enthalpy correction = 0.065462 kcal/mol ( 41.078 kcal/mol)
- model vibrational DOS enthalpy correction = 0.065500 kcal/mol ( 41.102 kcal/mol)
- vibrational DOS Entropy = 0.000018 ( 11.585 cal/mol-k)
- model vibrational DOS Entropy = 0.000019 ( 11.658 cal/mol-k)
- original gas Energy = -1035.813683 (-649982.894 kcal/mol)
- original gas Enthalpy = -1035.744446 (-649939.447 kcal/mol, delta= 0.000)
- unajusted DOS gas Enthalpy = -1035.744446 (-649939.447 kcal/mol, delta= -0.000)
- model DOS gas Enthalpy = -1035.744408 (-649939.423 kcal/mol, delta= 0.024)
- original gas Entropy = 0.000125 ( 78.674 cal/mol-k,delta= 0.000)
- unajusted DOS gas Entropy = 0.000126 ( 79.178 cal/mol-k,delta= 0.504)
- model DOS gas Entropy = 0.000126 ( 79.251 cal/mol-k,delta= 0.577)
- original gas Free Energy = -1035.781826 (-649962.904 kcal/mol, delta= 0.000)
- unadjusted DOS gas Free Energy = -1035.782066 (-649963.054 kcal/mol, delta= -0.150)
- model DOS gas Free Energy = -1035.782063 (-649963.052 kcal/mol, delta= -0.148)
- original sol Free Energy = -1035.787185 (-649966.266 kcal/mol)
- unadjusted DOS sol Free Energy = -1035.787425 (-649966.417 kcal/mol)
- model DOS sol Free Energy = -1035.787421 (-649966.415 kcal/mol)
DOS sigma = 100.000000
- vibrational DOS enthalpy correction = 0.065361 kcal/mol ( 41.014 kcal/mol)
- model vibrational DOS enthalpy correction = 0.065622 kcal/mol ( 41.178 kcal/mol)
- vibrational DOS Entropy = 0.000018 ( 11.441 cal/mol-k)
- model vibrational DOS Entropy = 0.000019 ( 11.943 cal/mol-k)
- original gas Energy = -1035.813683 (-649982.894 kcal/mol)
- original gas Enthalpy = -1035.744446 (-649939.447 kcal/mol, delta= 0.000)
- unajusted DOS gas Enthalpy = -1035.744548 (-649939.511 kcal/mol, delta= -0.064)
- model DOS gas Enthalpy = -1035.744286 (-649939.347 kcal/mol, delta= 0.100)
- original gas Entropy = 0.000125 ( 78.674 cal/mol-k,delta= 0.000)
- unajusted DOS gas Entropy = 0.000126 ( 79.034 cal/mol-k,delta= 0.360)
- model DOS gas Entropy = 0.000127 ( 79.536 cal/mol-k,delta= 0.862)
- original gas Free Energy = -1035.781826 (-649962.904 kcal/mol, delta= 0.000)
- unadjusted DOS gas Free Energy = -1035.782099 (-649963.075 kcal/mol, delta= -0.171)
- model DOS gas Free Energy = -1035.782076 (-649963.061 kcal/mol, delta= -0.157)
- original sol Free Energy = -1035.787185 (-649966.266 kcal/mol)
- unadjusted DOS sol Free Energy = -1035.787458 (-649966.438 kcal/mol)
- model DOS sol Free Energy = -1035.787435 (-649966.423 kcal/mol)
Normal Mode Frequency (cm-1) IR Intensity (arbitrary)
----------- ---------------- ------------------------
1 -0.000 0.994
2 -0.000 0.742
3 -0.000 0.126
4 0.000 0.198
5 0.000 0.066
6 0.000 0.014
7 90.940 0.278
8 167.140 1.995
9 254.880 0.132
10 323.090 1.819
11 446.400 13.086
12 661.700 66.862
13 791.170 22.837
14 839.950 3.951
15 944.690 6.536
16 968.860 33.759
17 1092.570 3.012
18 1176.580 4.613
19 1266.240 28.692
20 1299.890 9.479
21 1321.980 5.561
22 1489.310 4.348
23 1678.860 46.542
24 3101.310 4.404
25 3144.930 3.347
26 3184.670 0.514
27 3205.940 6.092
No Hindered Rotor Data
+-------------------------------------+
| Reactions Contained in the Database |
+-------------------------------------+
Reactions Containing INCHIKEY = UOORRWUZONOOLO-OWOJBTEDSA-N
Reactionid Erxn(gas) Hrxn(gas) Grxn(gas) delta_Solv Grxn(aq) ReactionType Reaction
All requests to Arrows were successful.
KEYWORDs -
reaction: :reaction
chinese_room: :chinese_room
molecule: :molecule
nmr: :nmr
predict: :predict
submitesmiles: :submitesmiles
nosubmitmissingesmiles
resubmitmissingesmiles
submitmachines: :submitmachines
useallentries
nomodelcorrect
eigenvalues: :eigenvalues
frequencies: :frequencies
nwoutput: :nwoutput
xyzfile: :xyzfile
alleigs: :alleigs
allfreqs: :allfreqs
reactionenumerate:
energytype:[erxn(gas) hrxn(gas) grxn(gas) delta_solvation grxn(aq)] :energytype
energytype:[kcal/mol kj/mol ev cm-1 ry hartree au] :energytype
tablereactions:
reaction: ... :reaction
reaction: ... :reaction
...
:tablereactions
tablemethods:
method: ... :method
method: ... :method
...
:tablemethods
:reactionenumerate
rotatebonds
xyzinput:
label: :label
xyzdata:
... xyz data ...
:xyzdata
:xyzinput
submitHf: :submitHf
nmrexp: :nmrexp
findreplace: old text | new text :findreplace
listnwjobs
fetchnwjob: :fetchnwjob
pushnwjob: :pushnwjob
printcsv: :printcsv
printeig: :printeig
printfreq: :printfreq
printxyz: :printxyz
printjobinfo: :printjobinfo
printnwout: :printnwout
badids: :badids
hup_string:
database:
table:
request_table:
listallesmiles
queuecheck
This software service and its documentation were developed at the Environmental Molecular Sciences Laboratory (EMSL) at Pacific Northwest National Laboratory, a multiprogram national laboratory, operated for the U.S. Department of Energy by Battelle under Contract Number DE-AC05-76RL01830. Support for this work was provided by the Department of Energy Office of Biological and Environmental Research, and Department of Defense environmental science and technology program (SERDP). THE SOFTWARE SERVICE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE SERVICE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE SERVICE.